|
Вверх
на главную страницу
История
Факты
Идеи
Люди
Библиография
Конференции
|
|
К вопросу о механизме действия сверхмалых доз Л.А. Блюменфельд
В последние годы в мировой научной литературе все чаще появляются работы, свидетельствующие о поразительных эффектах воздействия сверхмалых доз биологически активных веществ на протекание процессов в биологических объектах на молекулярном, субклеточном, клеточном уровнях и на уровне всего организма [1-6]. Подробный анализ этих и других публикаций можно найти в [E.B. Burlakova et. al, 1990]. Основной парадоксальный результат исследования действия сверхнизких концентраций биологически активных веществ можно сформулировать следующим образом. Активность веществ в области обычных молярных концентраций (10-7М и выше) меняется с изменением концентрации в полном соответствии с законами классической химии. При концентрациях 10-7-10-17 наблюдаются резкие отклонения от формального закона действующих масс, обнаруживаются максимумы активности при сверхнизких концентрациях.
При описании поведения природных объектов обычно вполне достаточно пользоваться стандартными характеристиками (термодинамическими параметрами и функциями). Для химических процессов, как правило, выбирают параметры: температуру Т, давление р, объем V, химический состав (число молей n1, n2,...ni). Иногда добавляют напряженность внешнего электрического и/или магнитного поля.
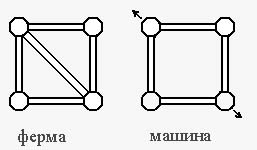
Есть еще одна особенность молекулярных конструкций, резко отличающая их от систем, не связанных с биологическими объектами. Физик, исследуя какую-либо систему, обычно задает два главных вопроса: как? и почему? Так, например, можно спросить: какова структура кристалла поваренной соли? И получить ответ: кристалл имеет кубическую симметрию, вокруг каждого иона натрия на вершинах куба расположены шесть ионов хлора, а вокруг каждого иона хлора -шесть ионов натрия. Далее физик спросит: почему кристалл NaCI построен именно так? И получит ответ: потому что такая структура отвечает минимуму энергии системы. Вопросы «почему?» вслед за первым можно задавать очень долго, пока не получишь ответ: «Это так, потому что это так», иначе говоря, пока не дойдешь до основных, недоказуемых постулатов, являющихся обобщением экспериментов и верных до тех пор, пока новые опыты не вступят с ними в противоречие.
Биологические системы и конструкции, сделанные ими, имеют цель [8]. Это значит, что, кроме указанных выше двух вопросов, можно задать вопрос для чего? или зачем? Так, например, можно спросить: «для чего молекула g-глобулина имеет такую-то структуру?» и получить ответ: «для того, чтобы выполнять иммунологическую функцию, что необходимо для выживания организма и вида». Цель биологическим конструкциям задает биологическая эволюция.
Все биологические объекты и их важнейшие компоненты кинетически неравновесны. Снятие кинетического запрета и изменение конструкции не требует изменения обмена энергии между системой и термостатом. Локальное возмущение, иногда энергетически или энтропийно ничтожное, может сделать кинетически доступными («отодвинуть защелку») другие конфигурации конструкции. Это требует корреляции между характеристиками возмущающего агента, например субстрата ферментативной реакции, и конструкции системы, например, конформации фермента.
Обратимся к первым попыткам рассмотрения возможных механизмов воздействия очень слабых возмущений на протекание химических реакций. Подходы, использованные в этих попытках, в значительной степени устарели (частично стали тривиальными, частично неверными), но некоторые, высказанные тогда идеи, оказались полезными в дальнейших работах. Солодовников исследовал спектры ЭПР некоторых анион-радикалов, в которых неспаренный электрон может быть локализован на двух ароматических кольцах, разделенных цепочкой из нескольких насыщенных связей. Было обнаружено, что даже при мостике из двух СН2-групп (анион-радикал [С6Н5-СН2-СН2 -С6Н5] -) неспаренный электрон одинаково взаимодействует с эквивалентными протонами обоих бензольных колец. Было высказано предположение о том, что заключение о наличии или отсутствии делокализации электрона (на языке химии — о наличии или отсутствии сопряжения) зависит от метода наблюдения: если частота u переноса электрона от одной потенциальной ямы (в данном случае бензольного кольца) к другой меньше обратного характерного времени метода наблюдения t (в данном случае значения постоянной сверхтонкой структуры в единицах времени), то наблюдатель зарегистрирует локализацию электрона в одном бензольном кольце. При u>1/t наблюдатель «увидит» электрон, делокализованный по двум бензольным кольцам. Идея Солодовникова была развита в нашей работе с Воеводским [18]. Слабые электронные взаимодействия, энергии которых меньше kT, но соответствующая частота которых превышает обратную величину характерного времени химической реакции, могут определять возможность ее протекания. В этой работе впервые шла речь об «управляющих взаимодействиях» в химии, характеризуемых не столько энергией, сравниваемой с kT, сколько временем, сравниваемым с характерным временем исследуемого процесса. В работе [18] было также высказано (правда, весьма нечетко) мнение, что такие слабые взаимодействия могут играть роль в ферментативном катализе, особенно в случае реакций, связанных с переносом электрона. В продолжение этой работы был проведен кванто-вомеханический анализ этих предположений, введены понятия делокализации электрона в стационарном и динамическом смысле и характерного времени метода измерения (последним может быть и химическая реакция) [19]. В простейшем случае, рассмотренном в [19], локализация электрона на время большее, чем характерное время элементарного акта реакции, приводит к появлению нестационарного квантового состояния с измененной реакционной способностью. В более сложных, в частности, биологических системах локальное возмущение, например, присоединение субстрата к активному центру фермента вызывает возникновение неравновесного состояния всего субстрат-ферментного комплекса.
Работа, получаемая за счет любого процесса, может быть связана как с изменением свободной энергии Гиббса, так и с изменением свободной энергии Гельмгольца (как правило, в биологических системах процессы идут практически в изобарных и изохорных условиях):  В интересующих нас биологических системах трансформацию энергии, обеспечивающую преодоление активационного барьера в ферментативных реакциях, в мембранных процесса, при фотофосфорилировании, мышечном сокращении и т.д., осуществляют устройства, принадлежащие к классу химических машин. Во всех этих случаях речь идет о сопряжении двух химических реакций: энергодонорной и энергоакцепторной. Энергия, освобождаемая в результате протекания энергодонорной реакции, тратится на реализацию энергоакцепторной реакции. Согласно (1), эти системы могут функционировать как энтропийные и энтальпийные машины. Начнем рассмотрение с энтропийных машин. В качестве примера такой «машины» можно взять сопряженный химический процесс, в котором сдвиг равновесия в одной реакции приводит к смещению равновесия в другой реакции благодаря наличию общего промежуточного продукта [20]: 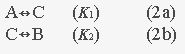 Отметим, что никакого переноса энергии между процессами (2а) и (2b) не происходит. Любой элементарный акт превращения А в С, С в А или В, В в А приводит либо к получению энергии из термостата, либо к отдаче энергии в термостат и не влияет непосредственно на возможность реализации других элементарных актов.
Как уже сказано выше, механизмы этого типа часто используют для описания энергетического сопряжения химических реакций: энергодонорная реакция (2а) обеспечивает протекание энергоакцепторной реакции (2b).
Оценим эффективность такого сопряжения. В качестве меры эффективности g выберем отношение числа результирующих элементарных актов n2 энергоакцепторного процесса С Следует обратить внимание на две особенности уравнения (4). Эффективность энергетического сопряжения по механизму Шилова [20] зависит в этом случае только от константы равновесия энергоакцепторной реакции К2 и может быть достаточно высокой (близкой к единице) лишь при больших значениях К2. Это, собственно, означает, что высокую эффективность превращения можно достигнуть лишь в тех случаях, когда «энергоакцепторная» реакция энергетически выгодна и, следовательно, не требует протекания энергодонорной реакции. Более подробно различные варианты такого энергетического сопряжения химических реакций рассмотрены в К2. Конечно, даже при К2 « 1 при достаточно длительном протекании энергодонорной реакции (2а) и сравнительно медленном протекании реакции (2b) можно накопить большой избыток промежуточного продукта С и перейти в стационарный режим с эффективностью g = 1. Стационарный режим реализуется при непрерывном поступлении исходного продукта А и отвода конечного продукта В после накопления сверхравновесной концентрации промежуточного продукта С. Однако многочисленные эксперименты свидетельствуют о том, что максимальная эффективность элементарного акта ферментативного превращения достигается практически мгновенно после начала энергодонорной реакции, даже если исходная концентрация промежуточного продукта С близка к нулю [22, 23].
Таким образом, трансдукция энергии между элементарными актами химических превращений не может быть непосредственно реализована за счет энтропийной компоненты изменения свободной энергии
системы. «Работает» только энтальпийная составляющая. Даже в типичных «энтропийных машинах», как например в концентрационных гальванических элементах, обязательным условием является превращение тепловой энергии термостата в «сохраняемую» форму потенциальной энергии, которая с помощью специального устройства может передаваться от одного участника процесса другому непосредственно, не диссипируя в термостат, если не принимать во внимание неизбежного трения или его аналогов, например, джоулевых потерь. Подробнее об этом см. в [22].
В основе рассматриваемого ниже подхода к выяснению механизма биокатализа лежат идеи о конфор-мационных изменениях молекулы фермента в ходе катализируемой реакции и о важной роли этих изменений в ферментативной активности. Эти идеи были выдвинуты много лет назад, обзор экспериментальных и теоретических работ в этом направлении, предшествующих появлению релаксационной концепции, можно найти, например, в [15]. Главным в подходе Сидоренко и Дещеревского было утверждение, что конформационная релаксация функционирующей молекулы фермента может играть важную роль в ферментативной активности. В то же время сам механизм превращения субстрата в продукт оставался в рамках классических представлений. Кон-формационные изменения в ходе релаксации фермента влияют на его активность, но не участвуют непосредственно в элементарном химическом акте. По-видимому, первое утверждение об отклонении элементарного каталитического акта ферментативной реакции от подходов классических термодинамики и кинетики было высказано в 1971 г. [26]. Было показано, что приложение основных постулатов теории активированного состояния к большинству ферментативных процессов приводит к физически бессмысленным значениям активационных параметров (энергии и энтропии активации). Функционирование фермента скорее аналогично работе механической конструкции, а не гомогенной каталитической химической реакции. В 1972 г. предложена самосогласованная феноменологическая теория ферментативного катализа [27, 28], а в 1976 г. В.М.Файн [29] разработал общую теорию кинетики химических реакций с участием высо-коупорядоченных молекулярных структур, которая базируется на основных постулатах релаксационной концепции. Файн дал самосогласованное описание одновременных изменений электронной и ядерной подсистем и получил уравнения, показывающие необычное поведение ферментативных процессов. Для высокоупорядоченных макромолекулярных систем в ходе безызлучательной релаксации избыток электронной энергии может быть трансформирован в энергию когерентных колебаний ядерной подсистемы без быстрой диссипации энергии в термостат.
Сформулируем основную идею релаксационной концепции ферментативного катализа. Речь идет не просто о том, что конформационная релаксация фермент-субстратного комплекса приводит к изменению каталитической активности фермента. Связывание субстрата с активным центром фермента инициирует конформационную релаксацию, действующую как движущая сила, которая перемещает химическую систему (молекула субстрата в каталитическом центре) вдоль координаты реакции. 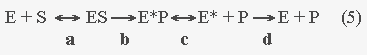 В этой предельно упрощенной схеме представлены два типа стадий: 1) быстрые обратимые стадии связывания субстрата S с ферментом Е (стадия а) и отщепления продукта Е*Р (с); 2) практически необратимые медленные релаксационные стадии (b) и (d). Каждая релаксационная стадия может быть записана в виде последовательности огромного числа обратимых элементарных актов (диссоциация или образование вторичных связей, поворот некоторых групп вокруг одиночных ковалентных связей и т.п.). После каждого элементарного акта системе представляется набор возможных и невозможных в данной мгновенной ситуации дальнейших элементарных актов, и движение по пути релаксации осуществляется методом бесчисленных проб и ошибок, с чем собственно и связана медленность релаксации. Детальное рассмотрение всего процесса можно найти, например, в [21].
Химическое превращение субстрата в продукт (S Из вышесказанного следует, что пути релаксации для прямой и обратной ферментативной реакции (упрощенная схема 5) могут быть различны. Экспериментальные данные и ссылки на соответствующие работы приведены в монографиях [1, 21, 22].
Рассмотрим одно из экспериментальных свидетельств возникновения неравновесных конформацион-ных состояний белков и роли их релаксации в функционировании ферментов [33, 34]. В эксперименте с использованием метода остановленной струи (stop-flow technique) следили одновременно за структурными изменениями белковой глобулы и за кинетикой катализируемого процесса в ходе прямой и обратной реакций, идущих при участии растворимой цитоплазматической малатдегидрогеназы и кофермента НАД : 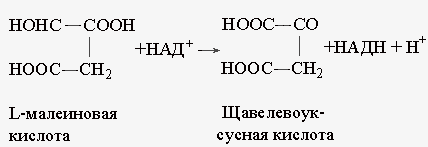 Соответствующие измерения проводили в ходе каждого ферментативного цикла (одного оборота фермента). Конформационные изменения в малатдегид-рогеназе регистрировали, измеряя среднее время жизни tf собственной флуоресценции триптофанового остатка. Известно, что этот параметр чувствителен к непосредственному окружению триптофана в белковой глобуле. Химическое превращение субстрата - малеиновой кислоты - детектировали по изменению окислительно-восстановительного состояния кофермента, измеряя оптическую плотность образца при 340 нм (максимум поглощения НАДН). Числа оборота фермента для прямой и обратной реакции равны, соответственно, 5,5*103 и 2,3*104мин-1. Это значит, что один акт прямой реакции занимает 10,9 мс, а обратной реакции — 2,6 мс. Кинетические характеристики прямой и обратной реакции не зависят от метода инициирования: добавление субстрата к смеси фермента и кофермента или добавление смеси кофермента с субстратом к раствору фермента. После инициирования прямой реакции структурно-чувствительный параметр tf уменьшается от исходного равновесного значения 3,1 мс до минимального значения ~ 2,4 мс через t = 6 мс. В конце первого цикла (t =11 мс) величина tf возвращается к исходному значению, а затем снова уменьшается с началом развития второго цикла. Восстановление НАД начинается только через 3-4 мс после инициирования реакции. Таким образом, образование продукта происходит только после завершения сравнительно значительных структурных изменений белковой глобулы. Совершенно иначе ведет себя обратная реакция. Протекание этой реакции практически не сопровождается изменениями tf , а образование продукта начинается сразу после смешивания участников реакции. Схемы, иллюстрирующие результаты эксперимента, приведены на рис. 2.
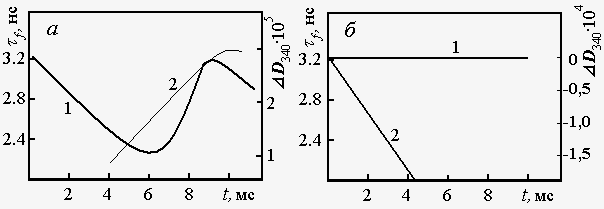
Полученные данные можно рассматривать как прямое доказательство того, что растворимая малат-дегидрогеназа катализирует прямую и обратную реакции, находясь в различных конформационных состояниях. Пути этих процессов не совпадают.
Релаксационная концепция биокаталитических процессов позволила предложить возможное объяснение действия сверхмалых доз биологически активных агентов на некоторые внутриклеточные процессы [35]. Наблюдаемые зависимости эффективности действия биологически активного вещества от его концентрации приводят к предположению, что при достаточно низких концентрациях возникает некий параметрический резонанс. Он определяется временными параметрами запускаемых биологически активными веществами внутриклеточных процессов и характерным временем доставки вещества (или продукта его взаимодействия с компонентами клетки) к специфическому месту внутри клетки (мембранный рецептор, активный или аллостерический центр фермента и т.п.). Можню предположить, что при сверхнизких концентрациях вещества лимитирующей стадией всего процесса будет диффузия его молекул из объема к поверхности клетки. Тогда характерное время доставки вещества к атомному центру t можно определить как обратную величину числа столкновений Z молекул вещества с . клеткой в единицу времени, т.е. t = Z -1 Пренебрегая размерами низкомолекулярного биологически активного вещества по сравнению с размерами биомишени (клетки), значение Z можно рассчитать с помощью известного уравнения Смолуховского:
В качестве характерного временного параметра внутриклеточного процесса, ответственного за наблюдаемый эффект, естественно выбрать время конфор-мационной релаксации макромолекулярных структур (ферментов, комплексов макромолекул, регуляторов биосинтеза, участков мембраны) после локального возмущения системы — присоединения субстрата, ингибитора или аллостерического лиганда к активному центру, редокс-изменения металла, ионизации кислотной группы и т.д. Для простоты дальнейшего изложения материала будем называть эту макромолекулярную структуру ферментом.
Итак, внешний агент — биологически активное вещество , поступающий в клетку со средней частотой Z, определяемой формулой (6), вызывает возникновение конформационно неравновесного состояния фермента, релаксирующего затем с характерным временем t к новому состоянию, равновесному для фермента с присоединенным агентом. На определенной стадии релаксации активность фермента экстремальна. С помощью уравнения (6) легко рассчитать молярные концентрации вещества при заданном радиусе мишени, соответствующие условию Z-1= t. Коэффициент диффузии D был выбран 5-10-10м2с-1 (сахароза в воде). Тогда получаем
Расчет показал, что для мишеней клеточных размеров (~ 1 мкм) можно ожидать появления экстремумов на концентрационной зависимости эффекта в интервале 10-11—10-15М. Это совпадает с данными в работе [1], посвященной влиянию ряда органических пероксидов на рост числа клеток в культуре клеток табака.
Литература
|
 В к числу элементарных актов n1 сопряженного энергодонорного процесса, А
В к числу элементарных актов n1 сопряженного энергодонорного процесса, А